
Como virus ARN, la replicación del genoma del SARS-CoV-2 es innatamente propensa a errores, de manera que se esperan mutaciones (1). De vez en cuando, una mutación proporcionará una ventaja adaptativa (como el aumento de la transmisibilidad o la infectividad a un huésped humano), y se seleccionará positivamente para en la población. Este proceso impulsa la evolución del genoma viral, y da lugar a la rica diversidad de cepas y linajes virales que vemos. Sin embargo, para el SARS-CoV-2, la inesperada aparición de tal diversidad desde finales de 2020 ha llevado a una ansiedad considerable en torno a la progresión de la enfermedad, y la eficacia de los diagnósticos y las vacunas para controlar su propagación.
El 22 de abril, Public Health England publicó la actualización 9 de las variantes de preocupación (COV) y variantes bajo investigación (VUI) del SARS-CoV-2 en inglaterra-Información técnica. Algunas buenas noticias es que no hay variantes conocidas de alta consecuencia (VOHC) en circulación hoy en día. Esta sesión informativa técnica es un gran recurso para comprender las variantes emergentes. Ha habido mucho reportado en las noticias (2,3) sobre una variante «Doble Mutante» en la India, por lo que estaba ansioso por leer más sobre esta cepa B.1.617.1: su perfil genómico, filogenia y caracterización biológica en la sesión informativa técnica.
Me sorprendió la familiaridad de las mutaciones en la variante B.1.617 India. Se trata del mismo «elenco de caracteres» que hemos visto en otras variantes de cepas preocupantes, mutaciones en las mismas posiciones de aminoácidos que siguen apareciendo, ya sea en diferentes combinaciones o como diferentes sustituciones de aminoácidos.
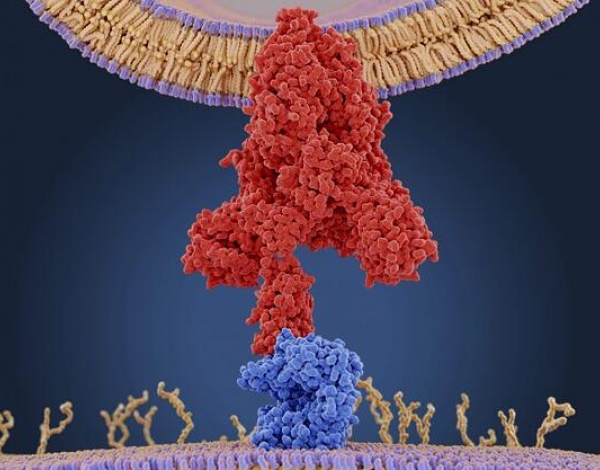
Figura 1: Modelo molecular de la proteína spike (S) (roja) unida a un receptor ACE2 (azul) en una célula humana. Imagen tomada de (4). Mientras que las mutaciones que definen el linaje surgen a lo largo del ARN genómico del SARS-CoV-2 que afectan a todas las proteínas virales, principalmente solo las mutaciones en el gen Spike hasta ahora han surgido repetidamente en linajes independientes. Las mutaciones E484 y L452 discutidas en la variante de la India B.1.617 están en la superficie de la proteína del punto que hace realmente el contacto con el receptor ACE2.
En el ejemplo de la variante de la India B.1.617:
- Mutación en la posición E484 del punto: Las mutaciones en esta posición fueron caracterizadas previamente en las variantes VOC-20DEC-02 (B.1.351 «Sudafricano») y VOC-21JAN-02 (P.1 «Japón/Brasil»). Las mutaciones E484K y, en menor medida, E484G/D/A/Y están implicadas en el escape antigénico, potencialmente a través de una mayor afinidad de unión al receptor ACE2 de la célula huésped humana (5). La nueva variante de la India VUI B1.617.1 tiene E484Q bastante que el E484K más extensamente estudiado, y todavía no se sabe cómo esta mutación afecta a ACE2 queta.
- Mutación en la posición L452 de Spike: L452R también se ve en la variante B.1.617 India y se encuentra en varias otras variantes previamente caracterizadas, incluyendo B.1.429 «Variante de California». Esta mutación también se asocia al escape antigénico de los anticuerpos monoclonales terapéuticos y de los antisueros convalecientes debido a la afinidad obligatoria aumentada del receptor. Algunos estudios sugieren que la afinidad de unión mejorada aumenta la capacidad de los virus para infectar las células humanas y, por lo tanto, mejora la transmisibilidad (6).
- Mutación en el sitio de escisión de la furina – Posición P681 de la espiga: La variante de la India B.1.617 contiene la mutación P681R del sitio de la hendidura de la furina, similar a P681H. Las mutaciones 681R/H se encuentran en múltiples variantes de linajes, como VOC-20DEC-01 (B.1.1.7 «variante del Reino Unido»), variantes de preocupación sars-CoV-2 y variantes bajo investigación VUI-21FEB-04 (B.1.1.318) y VUI-21FEB-01 (A.23.1). Tanto P681H como P681R optimizan la escisión de espigas por furina, lo que puede mejorar la transmisibilidad del virus.
Estos ejemplos en la variante B.1.617 India no son únicos. La posición de espiga Q677 ha mutado a H o P en al menos siete linajes diferentes, y los mutantes S-677 han aumentado la prevalencia una quintuplicación en todo el mundo (7). Del mismo modo, la deleción de espiga ΔH69/V70 está presente en al menos 28 linajes de SARS-CoV-2 (8). Ahora, las variantes de preocupación (VOC) del SARS-CoV-2 y las variantes bajo investigación (VUI) en inglaterra-Informe técnico 9 informes que además de la mutación muy común N501Y que definió inicialmente la «variante del Reino Unido» B.1.1.7, ahora hay una mutación N501T que se está monitoreando a partir de secuencias de Belo Horizonte, Brasil.
Las mismas mutaciones que aparecen una y otra vez sugieren una evolución convergente. La evolución convergente es cuando el mismo rasgo emerge en diferentes linajes independientes a lo largo del tiempo. La secuenciación del genoma a gran escala de virus de cientos de miles de pacientes (según lo catalogado en la base de datos GSAID) permite identificar estos patrones convergentes. La mayoría de las mutaciones son eventos puntuales que surgen en un paciente y se extinguen, pero algunos establecen nuevos linajes que se vuelven más frecuentes a medida que el virus logra replicar e infectar a muchas personas (7). Se ha documentado que los pacientes inmunodeprimidos producen múltiples mutaciones a lo largo del tiempo (9). Estos nuevos linajes se identifican como COV porque a menudo son responsables de las oleadas de COVID-19. Si las mutaciones en la misma posición de aminoácidos surgen repetidamente en todo el mundo y se vuelven más frecuentes, esta mutación muy probablemente codifica una adaptación que ayuda al virus a reproducirse y transmitirse. Al observar las mutaciones definitorias a través de los linajes de VOC podemos entender qué posiciones son importantes para la aptitud viral.
Reflexionando sobre las variantes discutidas en esta sesión informativa técnica, me sentí alentado por el hecho de que parece haber una lista limitada de variantes que afectan la transmisibilidad y virulencia viral. En lugar de un flujo interminable de nuevas variantes que son completamente impredecibles, la selección natural ya ha indicado las posiciones de aminoácidos que deben estar en nuestro radar para un seguimiento y vigilancia cercanos.
El monitoreo y la vigilancia de las variantes emergentes se pueden realizar utilizando ensayos de genotipado RT-PCR basados en sondas de alto rendimiento o utilizando la secuenciación del genoma completo (NGS). Independientemente del método, las pruebas de vigilancia de variantes requieren el uso de materiales de control de calidad para optimizar los ensayos, garantizar la sensibilidad y monitorear el control de calidad diario. LGC SeraCare produce materiales de referencia SARS-CoV-2 utilizando la tecnología patentada AccuPlex™. Estas partículas virales recombinantes están completamente encapsuladas como el patógeno SARS-CoV-2, por lo que deben pasar por un proceso de extracción antes de la detección de ácido nucleico, pero las partículas virales son defectuosas de replicación para un manejo seguro. Los materiales de referencia de la variante AccuPlex SARS-CoV-2 están actualmente en desarrollo. Los materiales contendrán el ARN genómico completo del SARS-CoV-2 y, para la primera liberación del producto, se centrarán en las mutaciones del gen S en tres variantes prominentes de preocupación (Tabla 1).
Tabla 1: Mutaciones y cepas variantes contenidas en el kit de material de referencia de la variante AccuPlex SARS-CoV-2. El kit tiene un vial para cada una de las tres variantes, así como un cuarto vial de control de tipo salvaje (secuencia de referencia NC_045512).
Con pruebas de alta calidad y eficientes para variantes, combinadas con una comprensión más profunda de las posiciones críticas de aminoácidos para aumentar la aptitud viral, las comunidades pueden reaccionar a las nuevas cepas de manera adecuada y, cuando sea necesario, implementar medidas de cuarentena de manera más efectiva.
Para obtener más información sobre el impacto de las variantes en las pruebas clínicas, vea el seminario web «Nuevos desafíos para el diagnóstico del SARS-CoV-2: pruebas de carga viral y genotipado de cepas variantes» presentado por el Dr. Nate Ledeboer y el Dr. Russell Garlick como parte de la serie de eventos virtuales de coronavirus LabRoots.